






Programming Projects: SCF with the JK object
Overview
In this project, you will learn how to write a restricted Hartree-Fock self-consistent field (SCF) solver as a plugin to the Psi4 electronic structure package. The solver will make use of the JK object in Psi4, which is capable of building coulomb-like (J) and exchange-like (K) matrices for arbitrary densities. The JK object can also make use of conventional, density-fitted, or Cholesky-decomposed electron repulsion integrals. If you would like to see how J and K can explicitly be built, see this tutorial.
For a tutorial that does not require the use of Psi4, see Daniel Crawford's SCF project (this project was adapted from that one). That project does not use density fitting, and all required integrals are read from text files so that your code can be written independently of any electronic structure package.
For additional information about the SCF procedure, I recommend Szabo and Ostlund's awesome (and cheap!) book Modern Quantum Chemistry or David Sherrill's online notes.
The C++ code for this SCF plugin can be found here.
Background:
Note: we use atomic units throughout this tutorial.
At the Hartree-Fock level of theory, the N-electron wave function is approximated as an antisymmetrized product of N one-electron functions called molecular orbitals (MOs). We call this type of wave function a Slater determinant, and the associated electronic energy can be expressed as
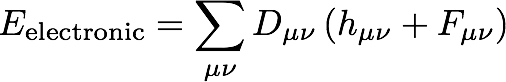
where D represents a density matrix (defined below), h represents the core Hamiltonian matrix, and F represents the Fock matrix. Here, Greek indices represent atomic orbital (AO) basis functions. The core Hamiltonian matrix contains integrals that represent the kinetic energy of an electron (T) and electron-nuclear potential energy (V):
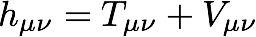
The matrix elements of the kinetic energy are integrals over the real-valued AO basis functions, χ, with the quantum-mechanical kinetic energy operator:
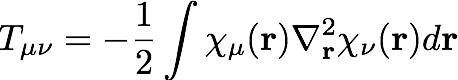
We should note here that these basis functions are not orthogonal, and we define the overlap matrix , S, as
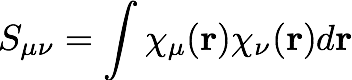
The matrix elements of the electron-nuclear potential energy are integrals over basis functions with the potential energy operator (Z/r):
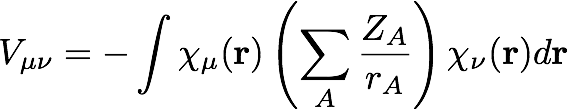
Here, there are multiple nuclei, labeled A, with nuclear charge ZA and a distance from the electron of rA. The Fock matrix is defined as
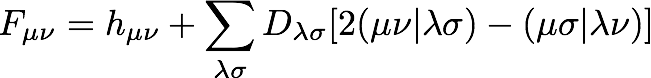
where the symbol, (μν|λσ) represents a two-electron repulsion integral, defined as
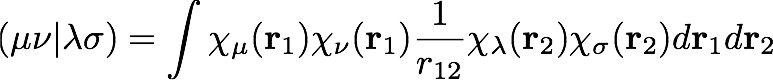
The actual working equations for Hartree-Fock theory are derived by considering a constrained minimization of the electronic energy with respect to the shape of the molecular orbitals, φ, which are expanded as a linear combination of atomic orbitals:
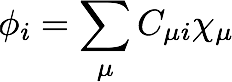
Using Lagrange's method of undetermined multipliers, the electronic energy is minimized with respect to the elements of C subject to the constraint that the molecular orbitals form an orthonormal set. Eventually, these considerations lead to an eigenvalue problem; the molecular orbitals are the eigenvectors of the Fock matrix, and the corresponding eigenvalues (which are the multipliers) are interpreted as the molecular orbital energies.
We now develop a computational procedure for determining the molecular orbitals and the electronic energy with the Psi4 electronic structure package.
Procedure:
Step 1. Create a new plugin
Create a new plugin, as described here. If you called your plugin "myscf," then all of your code can be placed inside plugin.cc. Open plugin.cc and locate the myscf(Options & options) function:
extern "C"
SharedWavefunction myscf(SharedWavefunction ref_wfn, Options& options)
{
int print = options.get_int("PRINT");
/* Your code goes here */
// Typically you would build a new wavefunction and populate it with data
return ref_wfn;
}
Like the comment says, your code goes there!
Before moving on, open the input file in your plugin directory (input.dat) and look at the molecule block
molecule {
O
H 1 R
H 1 R 2 A
R = .9
A = 104.5
# add this line!
symmetry c1
}
This plugin will not make use of point group symmetry, so you should add a line specifying that the computation be performed using C1 symmetry. Now, add a flag indicating that the computation will use density fitting techniques:
set {
# add this line!
scf_type df
basis sto-3g
}
For the remainder of this project, I've assumed that the rest of the input is unchanged. Keep this in mind when comparing to my results.
Step 2. Obtain 1-electron integrals
Now, close input.dat and open plugin.cc. In order to use Psi4's built-in integral libraries, matrix classes, etc., we must include the corresponding headers at the top of the file. Add the following headers:
#include "psi4/libmints/wavefunction.h" #include "psi4/libmints/mintshelper.h" #include "psi4/libmints/matrix.h" #include "psi4/libmints/vector.h" #include "psi4/libmints/basisset.h" #include "psi4/libmints/molecule.h" #include "psi4/lib3index/dftensor.h" #include "psi4/libqt/qt.h" // blas calls (in addition to those from Matrix/Vector classes) #include "psi4/libqt/qt.h" // jk object #include "psi4/libfock/jk.h"
We can now use Psi4's MintsHelper class to grab all of the one-electron integrals we'll need inside the function SharedWavefunction myscf(SharedWavefunction ref_wfn, Options& options).
// grab the one-electron integrals from MintsHelper:
std::shared_ptr<MintsHelper> mints (new MintsHelper(ref_wfn));
// one-electron kinetic energy integrals
std::shared_ptr<Matrix> T = mints->so_kinetic();
// one-electron potential energy integrals
std::shared_ptr<Matrix> V = mints->so_potential();
// overlap integrals
std::shared_ptr<Matrix> S = mints->so_overlap();
// build the core hamiltonian
std::shared_ptr<Matrix> h = (std::shared_ptr<Matrix>)(new Matrix(T));
h->add(V);
Note that all of the MintsHelper functions we call all have the prefix "so," which stands for "symmetry orbital." The SO basis is obtained by transforming the AO basis functions so that the SOs belong to a proper irreducible representation in the molecule's point group. If we are not using symmetry (which is the case here), then the AO and SO bases are equivalent.
Step 3. Initialize the JK object
At this point, we need to initialize the JK object, which is responsible for building our coulomb and exchange matrices. In order to initialize the JK object, we will need (1) the molecule object, (2) the primary basis set, (3) the auxiliary (or density-fitting) basis set.
// grab the molecule from the wavefunction that was passed into the plugin
std::shared_ptr<Molecule> mol = ref_wfn->molecule();
// get primary basis:
std::shared_ptr<BasisSet> primary = ref_wfn->get_basisset("ORBITAL");
// total number of basis functions
int nso = primary->nbf();
// get auxiliary basis:
std::shared_ptr<BasisSet> auxiliary = ref_wfn->get_basisset("DF_BASIS_SCF");
// total number of auxiliary basis functions
int nQ = auxiliary->nbf();
Note that we are using the strings "ORBITAL" and "DF_BASIS_SCF" when we grab the primary and auxiliary basis sets. What are these?
The primary basis set ("ORBITAL") should be set in the input file (e.g. "set basis sto-3g"). It was automatically set to STO-3G when you generated your plugin (check if you don't believe me!). The auxiliary basis set ("DF_BASIS_SCF") can be specified in the input file, but it was not automatically set when the plugin was created. Fortunately, since we requested that the scf_type be "DF", Psi4 is smart enough to pick a DF basis for you. If no "DF_BASIS_SCF" was specified in the input, then Psi4 picks the "JKFIT" variant of the primary "ORBITAL" basis that it deems appropriate. For the STO-3G basis, Psi4 will choose smallest possible "JK FIT" set, def2-svp-jkfit.
Moving on!
// JK object
std::shared_ptr<DiskDFJK> jk = (std::shared_ptr<DiskDFJK>)(new DiskDFJK(primary,auxiliary));
// memory for jk (say, 80% of what is available)
jk->set_memory(0.8 * Process::environment.get_memory());
// integral cutoff
jk->set_cutoff(options.get_double("INTS_TOLERANCE"));
// Do J/K, Not wK
jk->set_do_J(true);
jk->set_do_K(true);
jk->set_do_wK(false);
jk->initialize();
Here, we have set up the JK object to build the coulomb and exchange matrices, but we are not building any matrices associated with range-separated DFT (wK). Note that our JK object will use density fitting (DFJK), but you could have initialized the class to use conventional 4-index integrals or Cholesky-decomposed integrals. See the jk.h header file for more information.
Since this tutorial is designed for closed-shell systems, we should check to be sure that the number of electrons is even.
// use the molecule to determine the total number of electrons
int charge = mol->molecular_charge();
int nelectron = 0;
for (int i = 0; i < mol->natom(); i++) {
nelectron += (int)mol->Z(i);
}
nelectron -= charge;
// this code only works for closed shells
if ( nelectron % 2 != 0 ) {
throw PsiException("plugin myscf only works for closed shells",__FILE__,__LINE__);
}
// the number of doubly occupied orbitals (or alpha electrons)
int na = nelectron / 2;
Step 4. Symmetric orthogonalization
Oftentimes in electronic structure theory, we would prefer to work within a basis of orthonormal functions, which simplifies the math. However, our SO basis functions are not orthogonal. We can generate an orthogonal basis via Löwdin's symmetric orthogonalization. One first finds a transformation that diagonalizes the overlap matrix, S,
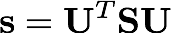
where s is a diagonal matrix of eigenvalues. We then construct the symmetric orthogonalization matrix by taking the inverse square root of the eigenvalues and backtransforming them to the original basis.
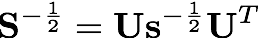
The following code snippet illustrates how to diagonalize the overlap matrix using Psi4's Matrix class. The back transformation is performed using a built-in function in the Matrix class.
// allocate memory for eigenvectors and eigenvalues of the overlap matrix
std::shared_ptr<Matrix> Sevec ( new Matrix(nso,nso) );
std::shared_ptr<Vector> Seval ( new Vector(nso) );
// build S^(-1/2) symmetric orthogonalization matrix
S->diagonalize(Sevec,Seval);
std::shared_ptr<Matrix> Shalf = (std::shared_ptr<Matrix>)( new Matrix(nso,nso) );
for (int mu = 0; mu < nso; mu++) {
Shalf->pointer()[mu][mu] = 1.0 / sqrt(Seval->pointer()[mu]);
}
// transform Seval back to nonorthogonal basis
Shalf->back_transform(Sevec);
Note that Shalf->pointer() gives you access to the underlying data in the Matrix class. Check your S-1/2 matrix using the Shalf->print() function.
Step 5. Guess orbitals, density, and energy
To obtain a set of guess orbitals and an initial density, we approximate the initial Fock matrix as the core Hamiltonian
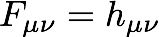
Next, use the symmetric orthogonalization matrix to bring this Fock matrix to a basis of orthonormal functions

Check your F' matrix.
We will now diagonalize this transformed Fock matrix to determine its eigenvectors (the molecular orbitals, C') and eigenvalues (the molecular orbital energies, ε)
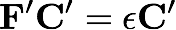
The matrix C' represents the molecular orbitals as a linear combination of orthonormalized basis functions, but what we really want is the linear combination of nonorthogonal AO (or SO) basis functions. We need to back transform one index of C' to the original AO basis
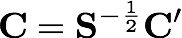
Since you are only transforming one index, don't use the Matrix class back_transform() call. You could use the Matrix class gemm() call
// allocate memory for SO->MO coefficients
std::shared_ptr<Matrix> Ca = (std::shared_ptr<Matrix>)(new Matrix(nso,nso));
// Find C = S^(-1/2)C'
Ca->gemm(false,false,1.0,Shalf,Fevec,0.0);
where Fevec is a Matrix containing the eigenvectors of F'. Alternatively, you could grab the appropriate pointers
double ** cp = Ca->pointer();
double ** sp = Shalf->pointer();
double ** fp = Fprime->pointer();
and transform that index using explicit loops. Check your MO coefficients.
Now, we define the density matrix as the contraction of the molecular orbital coefficients over the N/2 lowest-energy molecular orbitals (the doubly occupied orbitals):
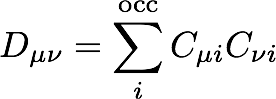
This density can then be used to evaluate the initial electronic energy and to obtain a new Fock matrix.
Check your density.
Step 6. The SCF procedure
You may have noticed that the Fock matrix depends on the density:
and the density is determined by diagonalizing the Fock matrix. If this seems a bit circular, you're right; we must determine the density for the system self-consistently. We repeatedly construct and diagonalize the Fock matrix until the electronic energy and density matrix cease to change. The self-consistent nature of the solution is the source of the name of the procedure: the Hartree-Fock self-consistent field procedure.
The SCF procedure is quite simple. Repeat Step 5, but use the correct Fock matrix (rather than the core Hamiltonian), until the changes in the energy and the density between iterations fall below some threshold.
Programming this expression is a little more manageable if we write the Fock matrix in terms of the coulomb and exchange matrices, J and K
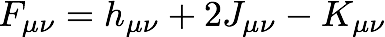
where
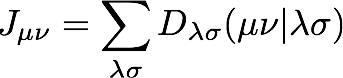
and
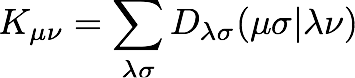
We will use the JK object to build the coulomb and exchange matrices. The JK object expects to build the density itself, so we need to pass the molecular orbital coefficients for the N/2 lowest-energy orbitals to it.
// grab occupied orbitals (the first na)
std::shared_ptr<Matrix> myC (new Matrix(Ca) );
myC->zero();
for (int mu = 0; mu < nso; mu++) {
for (int i = 0; i < na; i++) {
myC->pointer()[mu][i] = Ca->pointer()[mu][i];
}
}
// push occupied orbitals onto JK object
std::vector<SharedMatrix>& C_left = jk->C_left();
C_left.clear();
C_left.push_back(myC);
If we only push the left Ca matrix onto the JK object, it will assume that the right Ca matrix is the same. Note that the JK object could take different left and right Ca matrices. Also, it is possible to push more than one set of molecular orbital coefficients onto the JK object, in case you ever want to build multiple J and K matrices simultaneously. Now, actually building the J and K matrices is easy:
// form J/K
jk->compute();
// form F = h + 2*J - K
Fa->copy(jk->J()[0]);
Fa->scale(2.0);
Fa->subtract(jk->K()[0]);
Fa->add(h);
Check your coulomb, exchange, and Fock matrices from the first SCF iteration.
Step 7. Monitoring convergence
The SCF procedure is considered converged when the changes in the energy and density between iterations fall below some predefined thresholds. In Psi4, these convergence thresholds can be specified in the input file and obtained in your plugin from the "options" object:
// grab some input options
double e_convergence = options.get_double("E_CONVERGENCE");
double d_convergence = options.get_double("D_CONVERGENCE");
During the SCF procedure, we must monitor the change in the energy between iterations k-1 and k
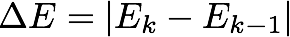
as well as the change in the density
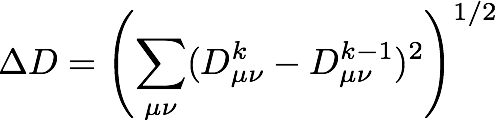
For the present molecule/basis set, the energy and density should converge to 8 decimal places in about 20 iterations:
Guess energy: -118.308230720196
==> Begin SCF Iterations <==
Iter energy dE dD
0 -73.196938802590 45.111291917606 1.897398367075
1 -74.939192979912 1.742254177322 0.112061053541
2 -74.944687307723 0.005494327811 0.028486807507
3 -74.945047846358 0.000360538636 0.010519702816
4 -74.945095717293 0.000047870934 0.004071261334
5 -74.945103245231 0.000007527938 0.001631352920
6 -74.945104500825 0.000001255594 0.000664268555
7 -74.945104714580 0.000000213755 0.000272761967
8 -74.945104751219 0.000000036639 0.000112497614
9 -74.945104757513 0.000000006294 0.000046510048
10 -74.945104758595 0.000000001082 0.000019254367
11 -74.945104758781 0.000000000186 0.000007976956
12 -74.945104758813 0.000000000032 0.000003306204
13 -74.945104758819 0.000000000006 0.000001370652
14 -74.945104758820 0.000000000001 0.000000568310
15 -74.945104758820 0.000000000000 0.000000235655
16 -74.945104758820 0.000000000000 0.000000097721
17 -74.945104758820 0.000000000000 0.000000040524
18 -74.945104758820 0.000000000000 0.000000016805
19 -74.945104758820 0.000000000000 0.000000006969
SCF iterations converged!
* SCF total energy: -74.945104758820
Note that the energy here is the total energy
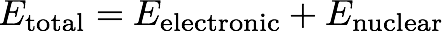
You can grab the nuclear repulsion energy from the molecule object:
double e_nuc = mol->nuclear_repulsion_energy({0.0,0.0,0.0});