New reactions of alkynes – alkynes as two functional groups in one package:
The alkyne, one of the simplest organic functional groups, has many interesting features associated with the presence of two types of bonds (π and σ) and the spatial orthogonality of the two π-systems. Furthermore, alkynes are in the same oxidation state as the −CH2C(O)– moiety and, thus, can be introduced in organocatalytic carbonyl pathways without the need for external redox agents.
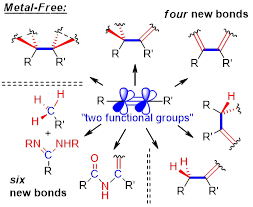
This combination of electronic features, together with the relatively high energy of alkynes, accounts for their rich chemistry and for the unique role of this functional group as a platform for the design of new reactions and for the discovery of new modes of chemical reactivity. In our lab, we have combined structural, energetic and stereoelectronic features of alkynes for the discovery of new cascade organic transformations. Examples include ionic chemistry of neutral hydrocarbons, preparation of radicals without radical initiators, generation of excited states without light, uncovering “1,2-dicarbene reactivity” of alkynes in “boomerang” radical processes, selective conversion of alkynes into carbonyl compounds, and full disassembly of the alkyne moiety.
Selected publications:
“Two functional groups in one package”: designing cascade transformations of alkynes.(Invited “Synopsis”) Alabugin, I. V.; Gold, B. J. Org. Chem., 2013, 78, 7777-7784. http://pubs.acs.org/doi/abs/10.1021/jo401091w.
Take a look at the unusual reactions of alkynes given below.
Alkynes as hidden 1,2-dicarbenes
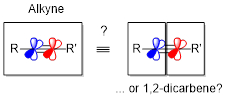
The relative stability of reactive intermediates that the energy gradient for the sp→sp2→sp3 cascades is favorable for the cationic and radical processes. Whereas strongly stabilizing catalysts/substituents are needed to compensate for the initial vinyl cation formation, vinyl radicals present an attractive compromise between reactivity and stability.
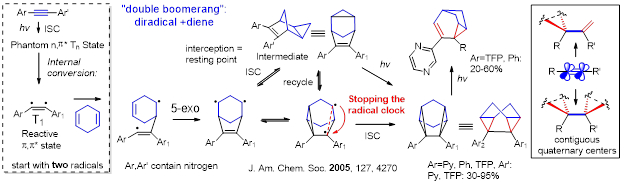
For unlocking the carbene-like reactivity patterns, vinyl radicals should undergo a reaction that initially translocates the radical center and then brings its attack back at the same atom where the radical was positioned initially. Below, we will show the two ways of launching such radical boomerangs: via alkene “boomerang” cycloadditions and via C − H translocation.
The cycloadditions, involved a “walk” of an initially formed radical center through an alkene before the “return” at the same carbon. Potential problem with this approach is that the penultimate cyclopropyl radical may undergo a fast “radical clock” ring opening. This diversion has to be eliminated, i.e., by running two of such clocks in the “opposite directions” (i.e., in a reaction of a diradical with a diene), so the two radicals “self-annihilate”.
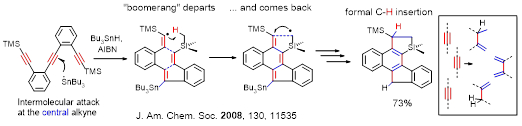
Carbene-like “insertion” of alkyne carbon into a C-H bond in the radical cascades involves sequential formation of C−H and C−C bonds at an alkyne carbon via a stepwise radical translocation.
Selected publications:
Zeidan, T.; Kovalenko, S. V.; Manoharan, M.; Clark, R. J.; Ghiviriga, I.; Alabugin I. V. “Triplet acetylenes as Synthetic Equivalents of 1,2-Dicarbenes. Phantom n,* State Controls Reactivity in Triplet Photocycloaddition”, J. Am. Chem. Soc. 2005, 127, 4270-4285. http://pubs.acs.org/doi/full/10.1021/ja043803l.
Alabugin, I. V.; Gilmore, K.; Patil, S.; Manoharan, M.; Kovalenko, S. V.; Clark, R. J.; Ghiviriga, I. Radical Cascade Transformations of Tris-(o-aryleneethynylenes) into Substituted Benzo[a]indeno[2,1-c]fluorenes, J. Am. Chem. Soc., 130, 2008, 11535-11545. http://pubs.acs.org/doi/abs/10.1021/ja8038213.
Alkynes as masked carbonyl equivalents (collaboration with S. F. Vasilevsky)
Because alkynes have the same oxidation level as carbonyl compounds, new alkyne cascade transformations can be engineered by unmasking the hidden “carbonyl nature” of alkynes. For example, addition of O- and N-nucleophiles, immediately transforms alkynes into enol and enamine derivatives.
The regiochemistry of the initial C-X attack chooses between the two latent carbonyl functionalities, rendering this step crucial for the success of cascade transformations. We are interested in unusual ways to control this selectivity. For example, one can leverage the stereoelectronic cyclization preferences to overcome polarization effects in the tug-of-war for regiochemical control in the alkyne-carbonyl conversion. For example, “anchoring” of multifunctional nucleophiles via 1,2-addition at the carbonyl of ketoacetylenes sets up an intramolecular “anti-Michael” attack which can be used for selective preparation of γ-dicarbonyls.
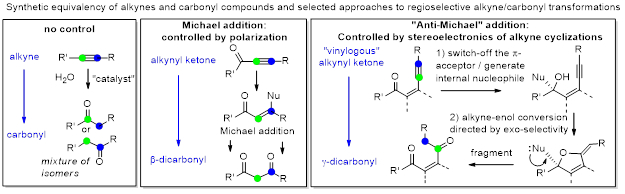
New cascade transformations can be developed via these approaches. For example, alkynes with acceptor substituents undergo regioselective reactions that reveal masked β-dicarbonyls. Subsequent retro-Mannich-like fragmentation leads to complete disassembly of the alkyne moiety via disproportionation.
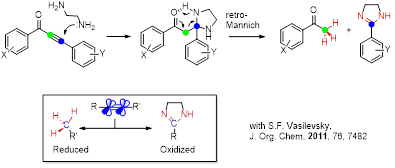
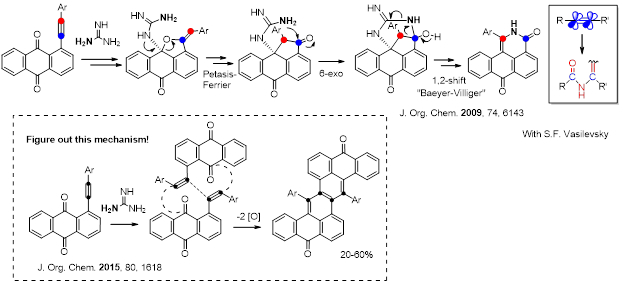
Alternatively, an “anchored” nucleophile initiates an “anti-Michael” cascade that inserts a nitrogen atom between the two alkyne carbons. This transformation leads to the formation of six new bonds at the two alkyne carbons with complete disassembly of the alkyne moiety. Note that the cascade is mediated by classic carbonyl chemistry, i.e., the fragmentation−recyclization analogues of the Petasis−Ferrier rearrangement and the [1,2]-shift converting the cyclic heminal into a lactam, similar to the final step of the Baeyer-Villiger oxidation.
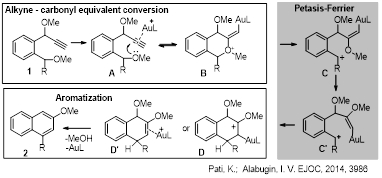
Au-catalyzed cycloisomerization of aryl propargyl ethers provides controlled transition from alkyne to carbonyl chemistry followed up by a Petasis–Ferrier rearrangement/aromatization cascade that opens synthetic access to substituted biaryls with a functionalized naphthalene subunit.
Selected publications:
Conformational Flexibility of Fused Tetracenedione Propellers Obtained from One-Pot Reductive Dimerization of Acetylenic Quinones. Vasilevsky, S. F.; Baranov, D. S.; Mamatyuk, V. I.; Fadeev, D. S.; Gatilov, Y. V.; Stepanov, A. A.; Vasilieva, N. V.; Alabugin, I. V. J. Org. Chem. 2015, 80, 1618-1631. http://pubs.acs.org/doi/abs/10.1021/jo502543b.
Synthesis of Substituted Biaryls Through Gold-Catalyzed Petasis-Ferrier Rearrangement of Propargyl Ethers. Pati, K.; Alabugin, I. V. Eur. J. Org. Chem. 2014, 19, 3986–3990. http://onlinelibrary.wiley.com/doi/10.1002/ejoc.201402469/abstract.
Baranov, D.; Gold, B.; Vasilevsky, S.; Alabugin, I. V. Divergent Cyclizations of 1-R-Ethynyl-9,10-anthraquinones: Use of Thiourea as a “S2-” Equivalent in an “Anchor-Relay” Addition Mediated by Formal C-H Activation. J. Org. Chem. 2013, 78, 2074-2082 (Special issue dedicated to H. E. Zimmerman). http://pubs.acs.org/doi/pdf/10.1021/jo302146r.
Baranov, D. S.; Vasilevsky, S. F.; Gold, B.; Alabugin, I. V. Urea as a Solvent and Reagent for the Addition/Cyclization/Fragmentation Cascades Leading to 2-R-7H-dibenzo[de,h]quinolin-7- one Analogues of Aporphinoid Alkaloids. RSC Adv., 2011, 1, 1745-1750. http://pubs.rsc.org/en/content/articlelanding/2011/ra/c1ra00622c.
Roy, S.; Davydova, M. P.; Pal, R.; Gilmore, K.; Tolstikov, G. A.; Vasilevsky, S. F.; Alabugin, I. V. Dissecting Alkynes: Full Cleavage of Polarized C≢C Moiety via Sequential Bis-Michael Addition/Retro-Aldol Cascade. J. Org. Chem., 2011, 76, 7482–7490. http://pubs.acs.org/doi/abs/10.1021/jo201259j.
Vasilevsky, S. F.; Baranov, D. S.; Mamatyuk, V. I.; Gatilov, Y. V.; Alabugin, I. V. An Unexpected Rearrangement which Disassembles Alkyne Moiety Through Formal Nitrogen Atom Insertion between Two Acetylenic Carbons and Related Cascade Transformations: New Approach to Sampagine Derivatives and Polycyclic Aromatic Amides, J. Org. Chem., 2009, 74, 6143–6150. http://pubs.acs.org/doi/abs/10.1021/jo9008904.
Vasilevsky, S. F.; Mikhailovskaya, T. F.; Mamatyuk, V. I.; Bogdanchikov, G. A.; Manoharan, M.; Alabugin, I. V. Tuning Selectivity of Anionic Cyclizations: Competition between 5-Exo- and 6-Endo-dig Closures of Hydrazides of o-Acetylenyl Benzoic Acids and Based-catalyzed Fragmentation/Recyclization of the Initial 5-Exo-Dig Products. J. Org. Chem. 2009, 74, 8106–8117. http://pubs.acs.org/doi/abs/10.1021/jo901551g.

