
NMR crystallography is an emerging field that utilizes NMR spectroscopy as the primary technique for predicting, refining, and validating crystal structures. 1H , 13C, and 15N chemical shifts are commonly used in NMRX protocols. Although easy to measure, they can be difficult and/or costly to calculate accurately using quantum chemical calculations. SSNMR of quadrupolar nuclei (I > 1/2) provides a powerful alternative, since acquisition of their spectra has been rendered relatively straightforward by modern magnets, new pulse sequences, and high magnetic fields. In addition, quadrupolar interactions are very sensitive to both short- and long-range structural changes and/or differences, and are also facile/inexpensive to calculate with quantum chemical methods.
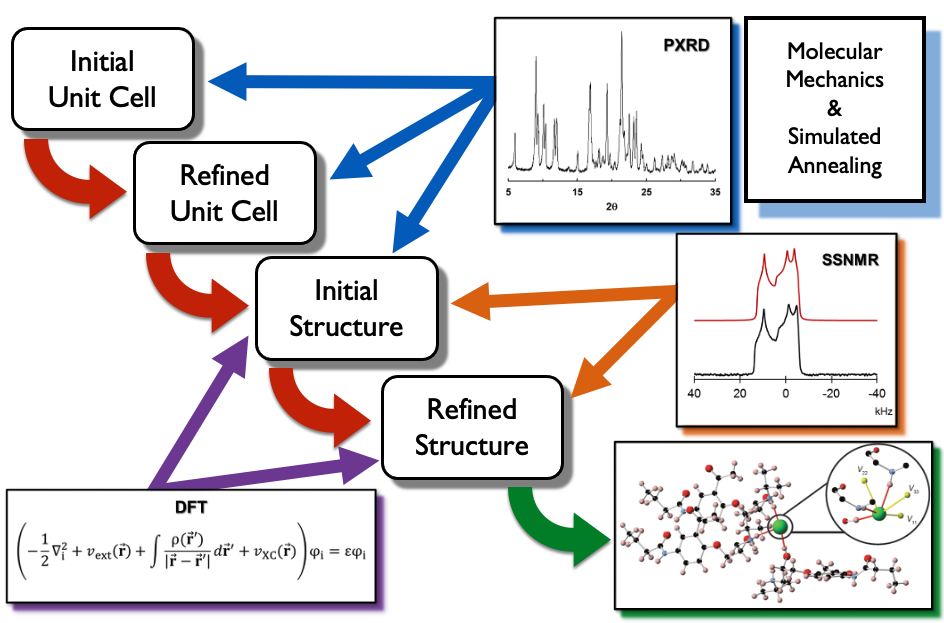
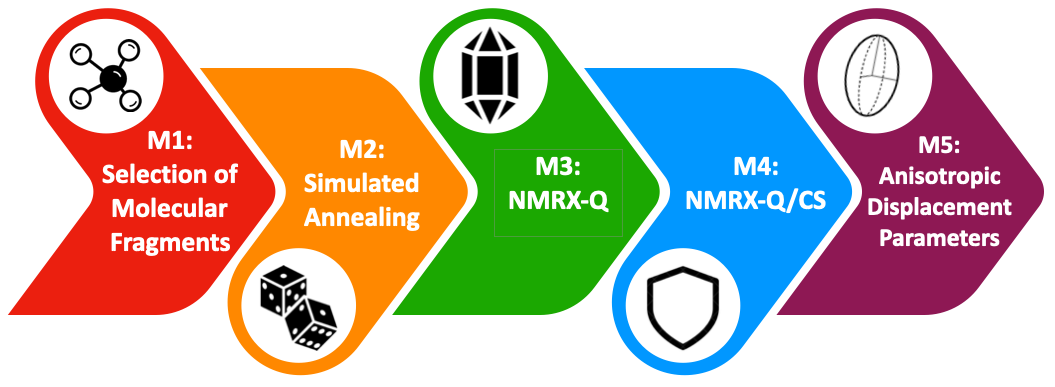
Our research group is developing an NMRX crystal structure prediction (NMRX-CSP) protocol based on SSNMR of quadrupolar nuclei:
• M1: Molecular fragments are built from gas phase calculations.
• M2: Fragments from M1 are fed into Monte Carlo simulated annealing routines, to generate thousands of candidate structures, which are filtered based on energies and unit cell parameters.
• M3: Hundreds of candidate structures from M2 are subjected to rapidly converging plane-wave DFT-D2* geometry optimizations, and then subjected to filters, which include comparisons of experimental and calculated 35Cl NMR quadrupolar parameters, reducing the number of candidate structures to under 10.
• M4: The remaining candidates from M3 are subjected to high-level plane-wave DFT-D2* geometry optimizations, and filtered on the basis of both quadrupolar and chemical shift parameters. This yields a final set of structures that should agree with known crystal structures.
• M5: New NMRX/MD methods are being explored for the purpose of determining anisotropic displacement parameters, similar to thermal ellipsoids from single-crystal X-ray structures.
• M1: Molecular fragments are built from gas phase calculations.
• M2: Fragments from M1 are fed into Monte Carlo simulated annealing routines, to generate thousands of candidate structures, which are filtered based on energies and unit cell parameters.
• M3: Hundreds of candidate structures from M2 are subjected to rapidly converging plane-wave DFT-D2* geometry optimizations, and then subjected to filters, which include comparisons of experimental and calculated 35Cl NMR quadrupolar parameters, reducing the number of candidate structures to under 10.
• M4: The remaining candidates from M3 are subjected to high-level plane-wave DFT-D2* geometry optimizations, and filtered on the basis of both quadrupolar and chemical shift parameters. This yields a final set of structures that should agree with known crystal structures.
• M5: New NMRX/MD methods are being explored for the purpose of determining anisotropic displacement parameters, similar to thermal ellipsoids from single-crystal X-ray structures.
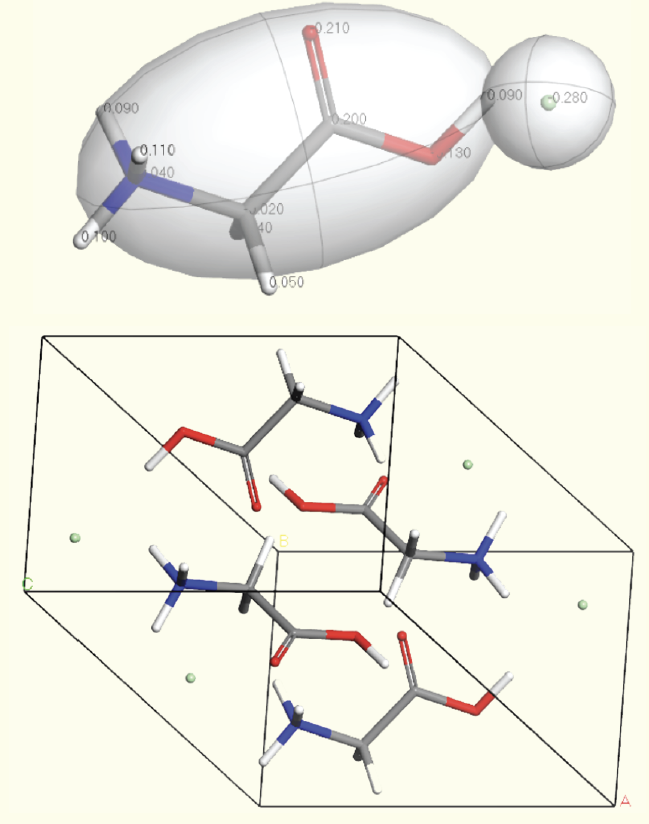
Pictured below are the gas phase fragments and final crystal structure of glycine HCl, as predicted by our method.
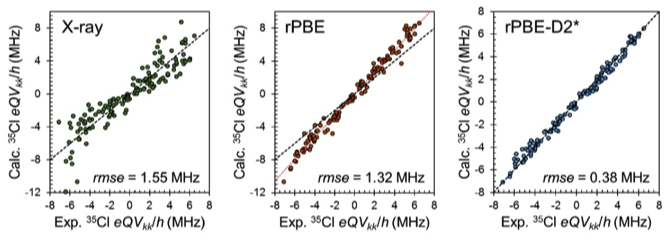
My group has been working on understanding the origins of NMR interactions and the relationships between NMR interaction tensors (chemical shielding and quadrupolar interactions) and molecular electronic structures in organic, biological, and inorganic systems. Much of our recent NMRX work is reliant upon the reparameterization of the reparameterizations of Grimme (D2 → D2*) and Tkatchenko-Scheffler (TS → TS*) force fields (best EFG/CS tensors) based on comparison of 150+ experimental and calculated EFG tensors (using CASTEP). Here, the adjusted dispersion parameter (d = 3.5) yields structures that provide the best agreement between experimentally measured and theoretically derived EFG tensors.
Selected computational and NMRX/CSP papers
- Holmes, S. T.; Hook, J. M.; Schurko, R. W. Nutraceuticals in Bulk and Dosage Forms: Analysis by 35 Cl and 14 N Solid-State NMR and DFT Calculations. Mol. Pharm. 2022, 19, 440–455. https://doi.org/10.1021/acs.molpharmaceut.1c00708.
- Holmes, S. T.; Engl, O. G.; Srnec, M. N.; Madura, J. D.; Quiñones, R.; Harper, J. K.; Schurko, R. W.; Iuliucci, R. J. Chemical Shift Tensors of Cimetidine Form A Modeled with Density Functional Theory Calculations: Implications for NMR Crystallography. J. Phys. Chem. A 2020, 124, 3109–3119. https://doi.org/10.1021/acs.jpca.0c00421.
- O’Keefe, C. A.; Mottillo, C.; Vainauskas, J.; Fábián, L.; Friščić, T.; Schurko, R. W. NMR-Enhanced Crystallography Aids Open Metal-Organic Framework Discovery Using Solvent-Free Accelerated Aging. Chem. Mater. 2020, 32, 4273–4281. https://doi.org/10.1021/acs.chemmater.0c00894.
- Holmes, S. T.; Vojvodin, C. S.; Schurko, R. W. Dispersion-Corrected DFT Methods for Applications in Nuclear Magnetic Resonance Crystallography. J. Phys. Chem. A 2020, 124, 10312–10323. https://doi.org/10.1021/acs.jpca.0c06372.
- Holmes, S. T.; Schurko, R. W. A DFT/ZORA Study of Cadmium Magnetic Shielding Tensors: Analysis of Relativistic Effects and Electronic-State Approximations. J. Chem. Theory Comput. 2019, 15, 1785–1797. https://doi.org/10.1021/acs.jctc.8b01296.
- Holmes, S. T.; Schurko, R. W. Refining Crystal Structures with Quadrupolar NMR and Dispersion-Corrected Density Functional Theory. J. Phys. Chem. C 2018, 122, 1809–1820. https://doi.org/10.1021/acs.jpcc.7b12314.
- Hildebrand, M.; Hamaed, H.; Namespetra, A. M.; Donohue, J. M.; Fu, R.; Hung, I.; Gan, Z.; Schurko, R. W. 35 Cl Solid-State NMR of HCl Salts of Active Pharmaceutical Ingredients: Structural Prediction, Spectral Fingerprinting and Polymorph Recognition. CrystEngComm 2014, 16, 7334–7356. https://doi.org/10.1039/C4CE00544A.
- O’Dell, L. A.; Schurko, R. W.; Harris, K. J.; Autschbach, J.; Ratcliffe, C. I. Interaction Tensors and Local Dynamics in Common Structural Motifs of Nitrogen: A Solid-State 14 N NMR and DFT Study. J. Am. Chem. Soc. 2011, 133, 527–546. https://doi.org/10.1021/ja108181y.
- Autschbach, J.; Zheng, S.; Schurko, R. W. Analysis of Electric Field Gradient Tensors at Quadrupolar Nuclei in Common Structural Motifs. Concepts Magn. Reson. Part A 2010, 36A, 84–126. https://doi.org/10.1002/cmr.a.20155.
- Widdifield, C. M.; Schurko, R. W. Understanding Chemical Shielding Tensors Using Group Theory, MO Analysis, and Modern Density-Functional Theory. Concepts Magn. Reson. Part A 2009, 34A, 91–123. https://doi.org/10.1002/cmr.a.20136.