Research







We develop new methods and algorithms for electronic structure theory. Click the links below to find out more!



Approximate coupled-cluster methods
We have developed a parametrized coupled-pair functional specifically designed to provide an accurate description of molecular interactions (PCPF-MI). Details can be found here. The method provides a description of nonbonded interactions that is superior to that of other coupled-pair methods of comparable cost.
The flexibility of familiar coupled-pair methods can be increased by introducing parameters into the renormalization denominator in the coupled-pair energy functional,
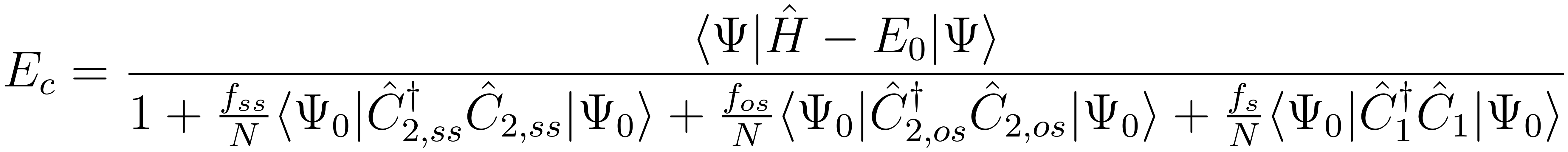
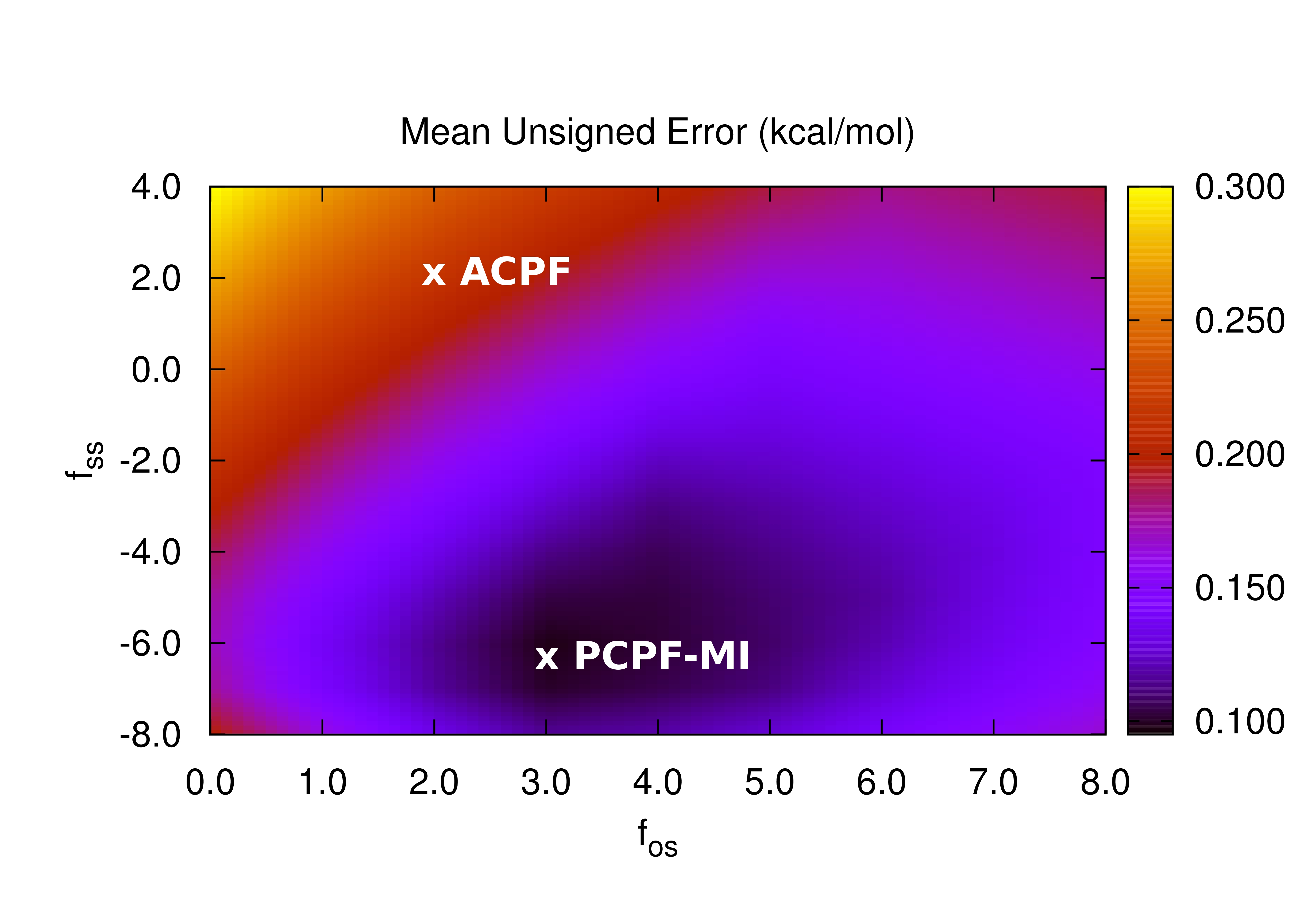
For the S22, HSG, S66 and A24 databases of van der Waals dimers, PCPF-MI computations yield interaction energies with MUEs of 0.326, 0.149, 0.214 and 0.044 kcal/mol, respectively, relative to benchmark computations.
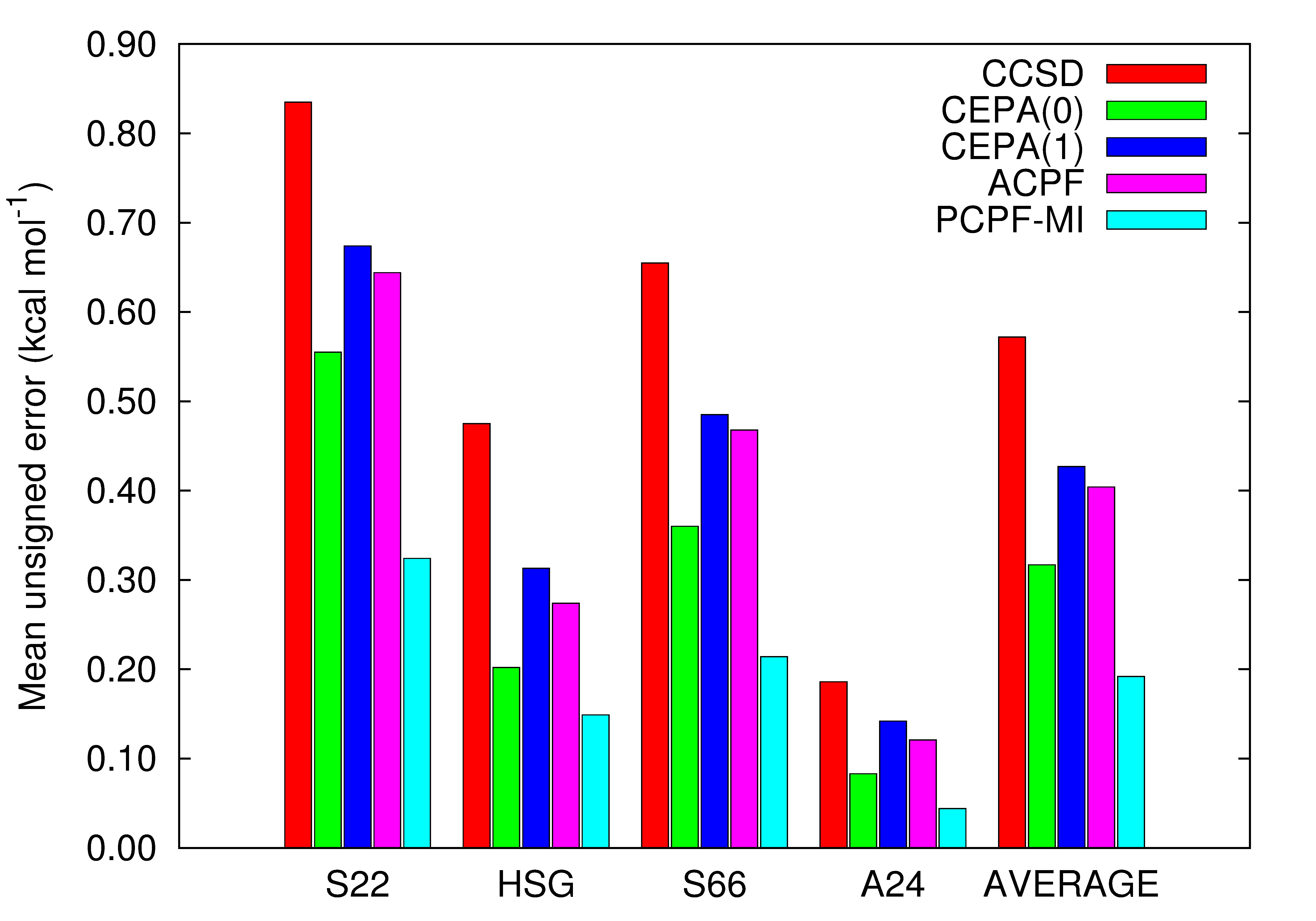
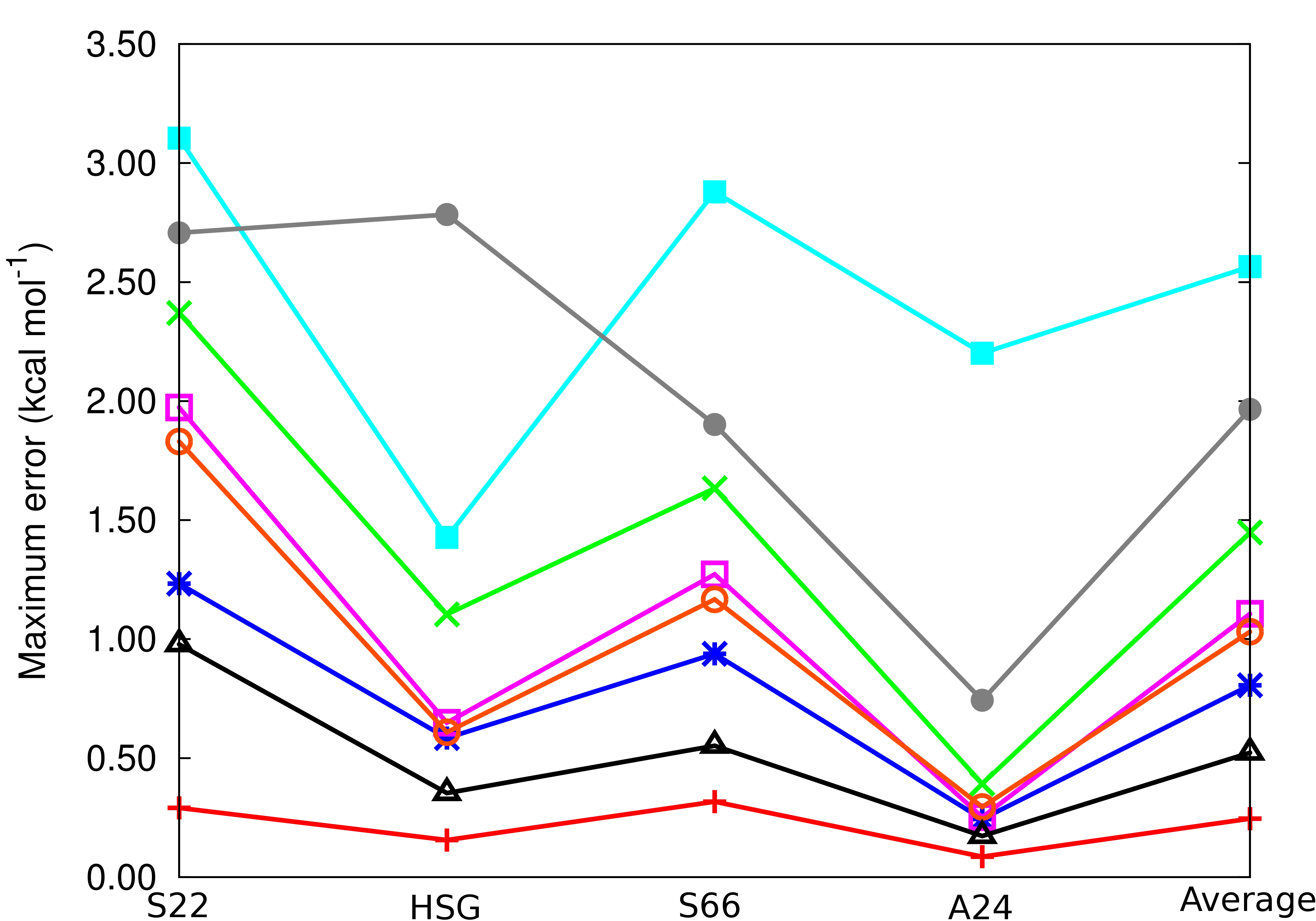
In addition to high accuracy, PCPF-MI is size extensive and yields energies that are stationary with respect to variations in all of its excitation coefficients. This last property facilitates the construction of density matrices and the evaluation of one and two-electron properties.