
Area 3: Nanoparticle-bioconjugates as analytical tools for sensing, imaging and diagnostics
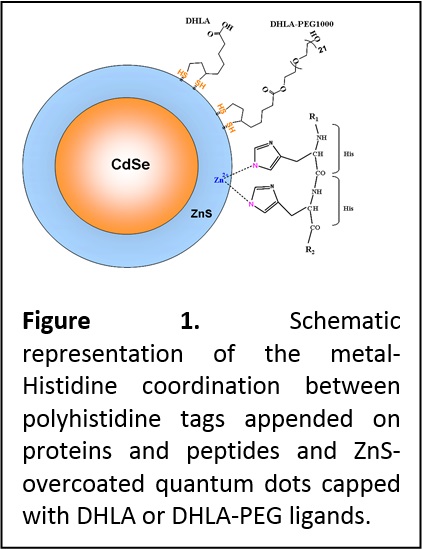
Development of QD-biomolecular assemblies. Early in the project, we developed a simple conjugation scheme to couple proteins and peptides to CdSe-ZnS QDs. The approach was based on the known affinity of polyhistidine tags to a whole array of metallic ions including Ni, Fe and Zn. We have shown that by engineering proteins and peptides with terminal polyhistidine sequences, metal-His driven self-assembly directly onto the Zn-rich QD surface can be realized (Figure 1). Furthermore, we showed that this self-assembly method could be applied to prepare a whole array of QD-bioconjugates, including QDs coupled to small and large size proteins, peptides and antibodies. We have also developed an understanding of the conditions under which metal-Histidine self-assembly can be realized along with the kinetics driving this process (J. Phys. Chem. C 2007, 111, 11528-11538. DOI: 10.1021/jp073550t). For example, we found that for QDs capped with the small size DHLA ligands, self-assembly can be realized with proteins and peptides alike as long as the histidine tag is exposed away from the receptor. When longer PEGylated ligands are used for surface capping, metal-affinity interactions can be applied only to polyhistidine-appended peptides, or to proteins engineered with a histidine tag appended at the end of a long linker; in both instances, the histidine tail is able to penetrate the ligand shell and interact directly with the metal-rich QD surface.
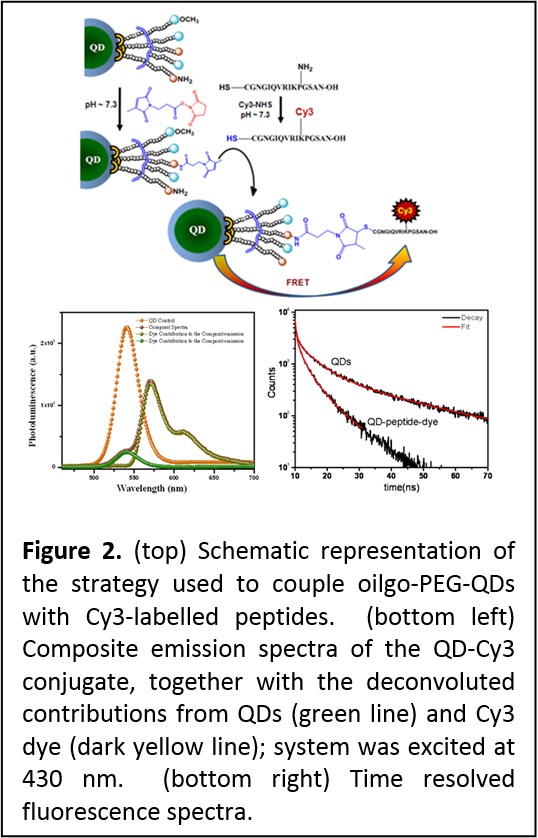
This particular result has motivated the development of our second generation DHLA-PEG-FN and OligoPEG ligands (see area 2). These allow conjugation based on covalent coupling (to target receptors) using EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) condensation, chemoselective ligation and maleimide-cysteine reaction (Figure 2). These coupling methods complement the metal-His conjugation strategy (Nat. Protocols 2009, 4, 424-436. DOI: 10.1038/nprot.2008.247; Small 2010, 6, 12731278. DOI: 10.1002/smll.201000279).
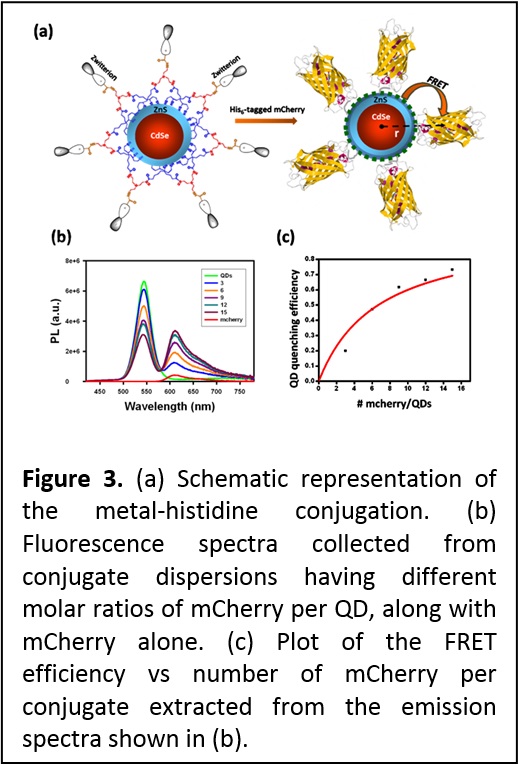
More recently we combined the great colloidal stability and compact size of QDs photoligated with zwitterion-terminated bis(lipoic) acid (see area 2) to self-assemble Qd-protein bioconjugates driven by metal-histidine coordination between the QDs and two different His-tagged proteins, maltose binding protein and fluorescent mCherry protein. We showed that these conjugates are biologically active. For instance, QD-MBP conjugates bind to amylase and are released by soluble maltose in affinity chromatography assay, while QD-mCherry conjugates engage in efficient energy transfer interaction as shown in Figure 3. The remarkable stability of QDs capped with these multi-coordinating and compact ligands combined with compatibility with metal-histidine conjugation, could be very useful for a variety of applications, ranging from protein tracking and ligand-receptor binding to intracellular sensing using energy transfer interactions (see J. Am. Chem. Soc. 2013, 135, 13786-13795. DOI: 10.1021/ja405010v).
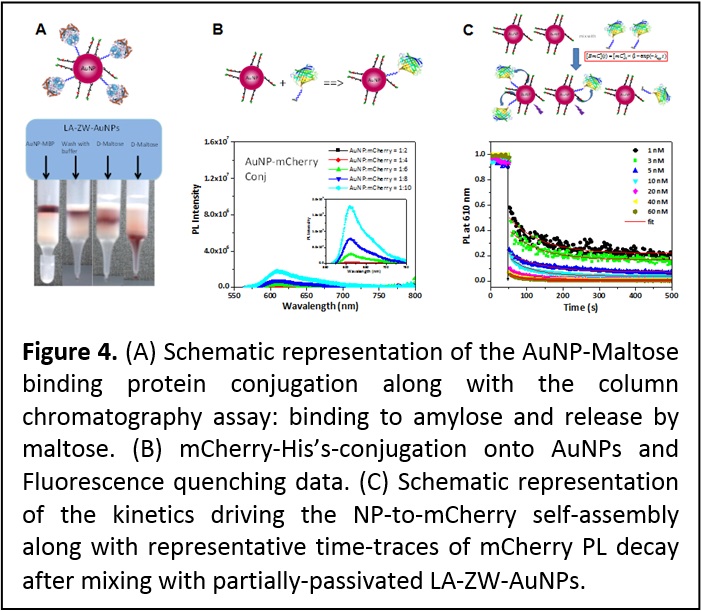
Self-assembly of protein onto metallic nanoparticles driven by metal-histidine coordination. Expanding the metal-histidine-driven conjugation scheme to other metal-rich nanoparticles (NPs) such as those made of Au and Ag would be of great interest to researchers actively seeking effective means for interfacing inorganic nanostructured materials with biology. To test this idea we investigated the metal-affinity self-assembly between AuNPs and two engineered proteins, a His7-appended maltose binding protein (MBP-Hisn) and a fluorescent His6-terminated mCherry protein as shown in Figure 4. In particular, we probed the influence of the capping ligand affinity to the nanoparticle surface, its density, and its lateral extension on the AuNP-protein self-assembly. Affinity gel chromatography was used to test the AuNP-MPB-His7 self-assembly, while NP-to-mCherry-His6 binding was evaluated using fluorescence measurements. We also assessed the kinetics of the self-assembly between AuNPs and proteins in solution, using time-dependent changes in the energy transfer quenching of mCherry fluorescent proteins as they immobilize onto the AuNP surface (Figure 4). This allowed determination of the dissociation rate constant, Kd-1 ~ 1-5 nM. Furthermore, a close comparison between the protein self-assembly onto AuNPs or onto QDs provided additional insights into which parameters control the interactions between imidazoles and metal ions in these systems (ACS Nano 7, 10197–10210 (2013). DOI: 10.1021/nn404479h).
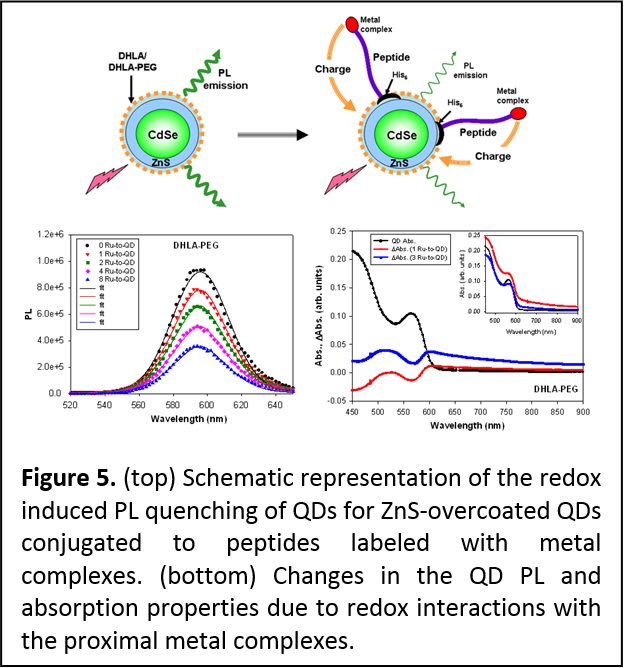
Interaction between QDs and redox active complexes. Colloidal quantum dots (QDs) have a large fraction of their atoms arrayed on their surfaces and are capped with bifunctional ligands. This makes their photoluminescence highly sensitive to potential charge transfer to, or from, the surrounding environment. We coupled metal complexes with ionization potentials comparable or much larger than that of the hydrophilic DHLA- and DHLA-PEG-QDs. The metal-His self-assembly was used to control the separation distance and the number of metal complexes bound to an individual QD. We found that quenching of the QD emission takes place only for complexes whose oxidation levels are either lower or equal to that of the QDs; rates of PL quenching also tracked the number of metal complexes arrayed around a single nanocrystal. In addition, partial bleaching of the absorption was measured for the QD-metal complex assemblies (Figure 5). We attributed these results to charge transfer from the metal complex to the QD surface state which, in addition to altering the exciton recombination and PL emission, produces changes in the absorption spectra. These changes were also found to depend on the nature of the surface ligands (neutral DHLA-PEG versus charged DHLA) (J. Am. Chem. Soc. 2008, 130, 16745-16756. DOI: 10.1021/ja805456x). QDs coupled to complexes exhibiting higher oxidation potentials provided higher barrier for electron transfer to the QDs and produced no PL quenching and no change in the QD absorption features.
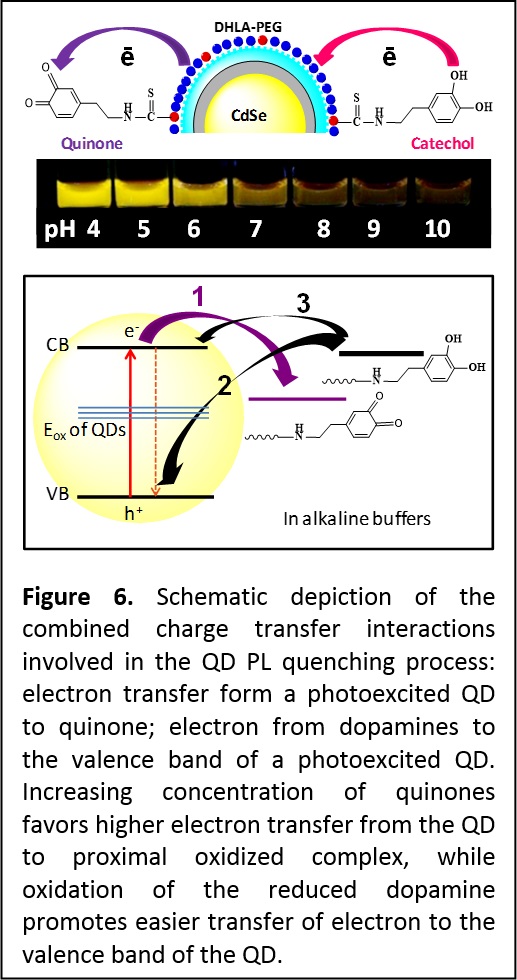
More recently we expanded these findings and used dopamine, a redox complex with strongly pH-dependent oxidation potential; dopamine also has some important biological implications centered on brain operation and hypertension. Dopamine was conjugated onto the QDs via two different strategies. One strategy relied on attaching dopamine to a polyhistidine-modified peptide, and then assembling the peptide-dopamine complex onto PEGylated CdSe-ZnS QDs via metal-His coordination. The other strategy used covalent conjugation to attach the dopamine directly onto the PEG ligand on the nanocrystals. Here, we used pH-insensitive ZnS-overcoated CdSe QDs, rendered water-compatible using poly (ethylene glycol)-appended dihydrolipoic acid (DHLA-PEG), where a fraction of the ligands was amine-terminated to allow for controlled coupling of dopamine-isothiocyanate onto the nanocrystal. We probed the effects of changing the density of dopamine and the buffer pH on the fluorescence properties of these conjugates. Using steady-state and time-resolved fluorescence we measured a pronounced pH-dependent photoluminescence (PL) quenching for all QD-dopamine assemblies (Figure 6). We found that several parameters affect the PL loss, including the number of dopamines per QD-conjugate, the environment pH (quenching efficiency is substantially increased in alkaline buffers), and oxygen dissolved in the buffer. We attribute these findings to charge transfer interactions between QDs and primarily two forms of dopamine: the reduced catechol and oxidized quinone (Figure 6). As the pH of the dispersions is changed from acidic to basic, oxygen-catalyzed transformation progressively reduces the dopamine potential for oxidation and shifts the equilibrium towards increased concentration of quinones. Thus, in a conjugate a QD can simultaneously interact with quinones (electron acceptors) and catechols (electron donors), producing pH-dependent PL quenching combined with shortening of the exciton lifetime. This also alters the recombination kinetics of the electron and hole of photoexcited QDs. Transient absorption measurements that probed intraband transitions supported these findings where a simultaneous pronounced change in the electron and hole relaxation rates was measured when the pH was changed from acidic to alkaline. We found that such QD-dopamine assemblies could be used to sense in vitro and intracellular pH changes (Nat. Mater. 2010, 9, 676–684. DOI:10.1038/nmat2811 and J. Am. Chem. Soc. 2012, 134, 6006−6017. DOI: 10.1021/ja300724x).
Energy transfer-based sensing. We have investigated energy transfer interactions between QDs as donors and acceptors and designed sensing constructs based on FRET (fluorescence resonance energy transfer). We have, for example, shown that the point dipole approximation applies to luminescent QDs and that energy transfer with QDs (e.g., QD-dye pairs) is governed by the Förster dipole-dipole mechanism (J. Am. Chem. Soc. 2004, 126, 301-310 (2004). DOI: 10.1021/ja037088b; J. Phys. Chem. C 2009, 113, 18552–18561. DOI: 10.1021/jp9060329). We have also shown that QD donors offer several unique features not available to molecular organic dyes, namely tunable excitation of the donor-acceptor pair, reduced direct excitation of the acceptor (e.g., dye), and the ability to increase the FRET rates by arraying multiple acceptors around a single QD donor. We have further developed several sensing configurations based on QDs and FRET. These include the use of competitive sensing combined with QD-protein conjugates to detect sugars and soluble TNT, and use of QD-peptide assemblies as substrates to detect proteolytic enzyme activity. More recently we explored energy transfer between QDs and proximal dyes under two-photon excitation, and demonstrated that in this format QDs provide additional unique advantages due to their very large two-photon action cross section compared to organic dyes. For instance, with two-photon excitation, dye contribution resulting purely from FRET can be generated and easily detected. This is particularly advantageous for intracellular sensing and imaging (Adv. Mater. 2007, 19, 1921-1926. DOI: 10.1002/adma.200602036).
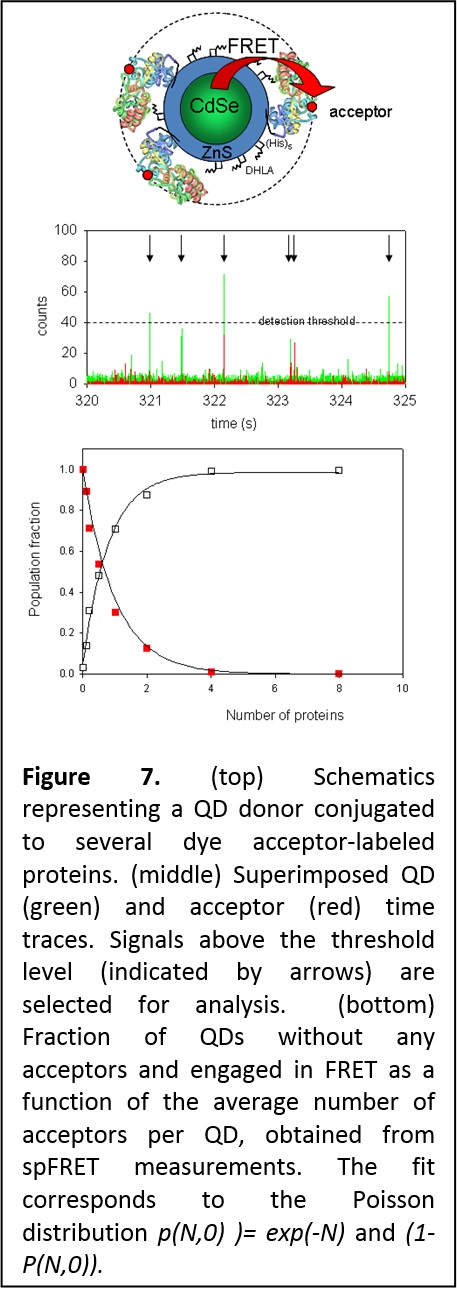
Heterogeneity of QD-protein conjugates: Single particle FRET with QDs donors. We used solution-phase single molecule/particle FRET (spFRET) to characterize the heterogeneity of self-assembled QD-bioconjugates and to gain unique insight into the distribution in conjugate valence. Heterogeneity in conjugate valence is an inherent property of nanoparticle-conjugates because of the large surface area of these nanoparticles, independent of the conjugation strategy or nanoparticles used. We used spFRET to confirm the heterogeneous nature of QD-protein self-assemblies, and further demonstrated that the distribution in the valence is governed by Poisson statistics (Figure 7). This is a crucial finding for these systems, as often a controlled valence may be desired for certain specific applications (J. Am. Chem. Soc. 2006, 128, 15324-15331. DOI: 10.1021/ja0657253).
We also applied spFRET to immobilized QD-dye conjugates to confirm and complement spectrally resolved energy transfer observed for ensemble measurements. Spectrally-resolved FRET is a unique property of QD donors and is due to a combination of broad dye absorption and the extremely narrow emission (15 nm) of individual QDs. It is observed when the narrow QD emission overlaps with either the blue or red regions of the broad acceptor absorption. It can produce a wavelength-dependent rate of FRET combined with either red shift in the PL spectrum if the latter coincides with the red shoulder of the dye absorption spectrum, or a blue shift if it coincided with the blue shoulder of the dye absorption.
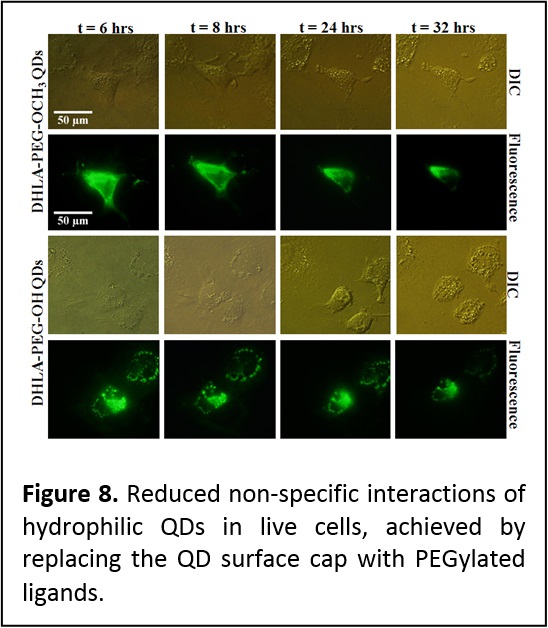
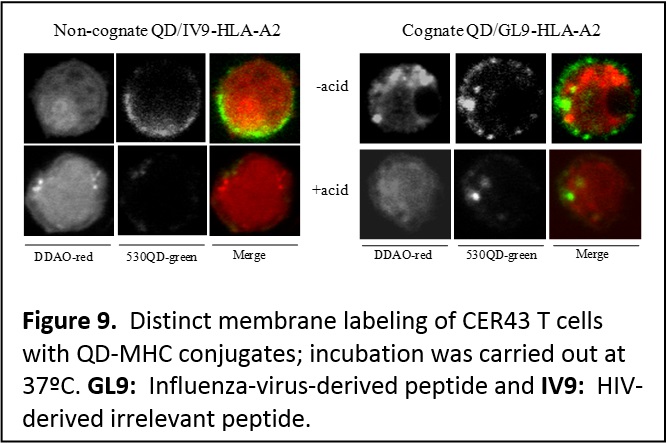
Probing cellular and membrane specific interactions using QD-bioconjugates. We have shown that introducing PEGylated function into the capping ligands provide QDs that exhibit reduced non specific interactions and aggregation in live cells following microinjection into eukaryotic live cells (Figure 8) (J. Mater. Chem. 2008, 18, 4949-4958. DOI: 10.1039/b810488c). In collaboration with researchers at the Kimmel Cancer Institute (Y. Sykulev) and New York Univ. Medical School (M. Dustin), we employed QDs conjugated to the major histocompatibility complex to probe the interactions that occur at the membranes of Cytotoxic T lymphocytes (CTL or T cells), and delineate the contributions of co-receptor proteins to the T cell recognition of target infected cells. This provided a better understanding of the mechanisms that drive the high sensitivity of T cells. Due to its rather large size, a QD can simultaneously accommodate several protein receptors (single or multiple types of proteins), and this could provide additional benefits for probing specific processes occurring at the cell membrane. We utilized QDs self-assembled with multiple copies of His-appended MHC-I (major histocompatibility class I) proteins to examine the recognition by antigen-specific T cell receptors (TCRs) on anti-virus CD8-T cells. These self-assembled QD-pMHC-I conjugates provided two unique features: [1] a structure where orientation and spacing between proximal complexes resemble what live T cells encounter in a cell system, and [2] the ability to titrate the number of virus-derived (antigenic) and other (non-antigenic) pMHC-I complexes within a conjugate, which also mimics what a target infected cell can present on its surface. In particular, we investigated the kinetics of T cell stimulation in the presence of different combinations of intact and mutated MHC-I proteins and antigenic and non-antigenic peptides conjugated to the nanocrystals (Figure 9). We showed that strong multivalent interactions between the MHC-I and CD8 co-receptor protein (characteristic of certain types of T cells) promote/facilitate an additional and effective contribution of non-antigenic pMHC-I to the recognition of antigenic pMHC-I by T cell receptors. Such cooperation spreads the recognition signal from a few engaged to several additional TCRs (Proc. Natl. Acad. Sci. USA (PNAS) 2006, 103, 16846-16851. DOI: 10.1073/pnas.0607771103).
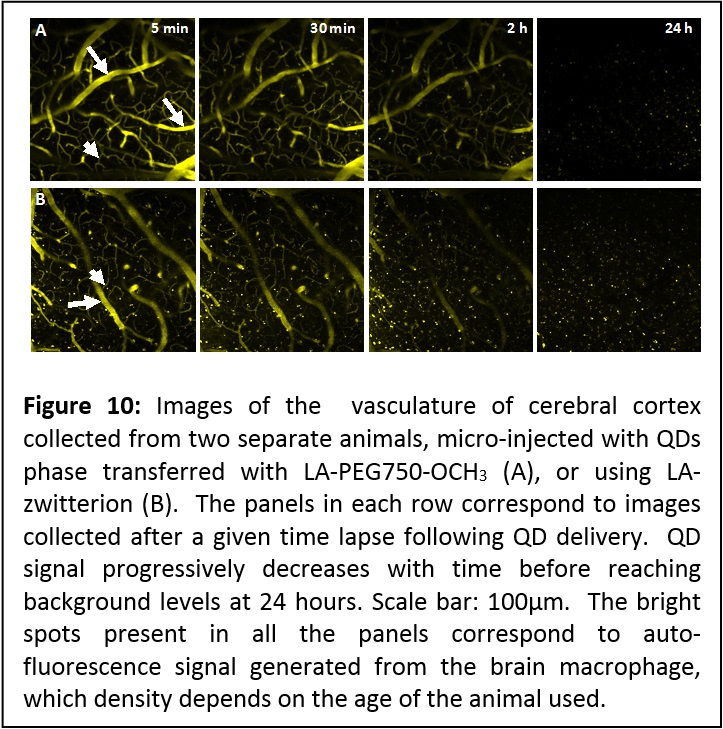
Application of these QDs to imaging the brain of live mice. We have exploited the high two-photon fluorescence properties of QDs to image the brain vessels (including capillaries) of live mice using QDs phase-transferred to buffer media (using a newly developed strategy based on UV-induced ligand exchange, see area 2) in the presence of LA-PEG-OCH3 or LA-zwitterion ligands. The fluorescence images show well resolved capillary structures with a submicron resolution (see Figure 9). The vasculature could be effectively imaged up to ~200 μm deep (from the surface). In particular, the QDs remained visible in the circulating blood for up to 2 hours without any visible self aggregation or blockade in microcapillaries. The signal decreased to background levels within 24 hours, which suggests that these QDs do not inherently partition into brain tissue or endothelial cells, and were progressively cleared from the blood circulation, likely due to renal filtration. Indeed, we observed increased fluorescence in mouse urine, 2 hours after injection. We should emphasize that the ability to image the blood brain capillary for such extended period of time constitutes a very promising capability, as it permits investigation of the structure and integrity of the blood capillaries in the brain of live animals under different conditions (structure < 10 μm in diameter, see Figure 10). These hydrophilic QDs provide a high signal-to-noise ratio, and due to a combination of their nanometer size and relatively inert coating, have significantly extended circulating half life, allowing effective imaging of the capillaries over several hours (for more details see report in J. Am. Chem. Soc. 2012, 134, 16370–16378. DOI: 10.1021/ja306621n).
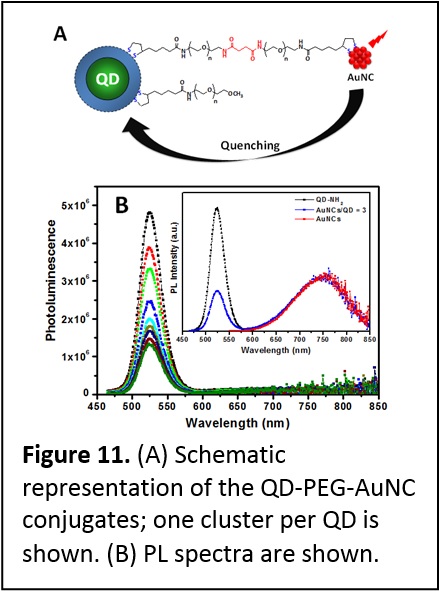
Quenching of QD Emission by Fluorescent Gold Clusters. We have engineered a unique model where fluorescent gold nanoclusters (AuNCs) were coupled to CdSe-ZnS QDs covalently (Figure 11). This system allowed us to understand the interactions that control the energy transfer process between QDs or dye and metallic nanoparticles. These interactions were probed using steady state and time-resolved fluorescence measurements. We systematically investigated the spectral overlap effects and separation distance on the quenching of QD photoemission by proximal AuNCs, using three different size QDs with distinct emission spectra and a variable length polyethylene glycol bridge. We found that the QD photoluminescence quenching efficiency depends on the spectral overlap and separation distance, with larger quenching efficiencies than what would be expected for a QD-dye pair with similar overlap. Furthermore, despite the large losses in QD PL, we found no resonance enhancement in the cluster emission for any of the sample configurations used. These results indicate that the mechanism driving the quenching by metal clusters shares an important feature (namely dependence on the spectral overlap) with the Förster dipole-dipole coupling at the heart of fluorescence resonance energy transfer (FRET) which has been widely validated for dye-dye and QD-dye assemblies. These results prove that the energy losses induced by metal nanostructures are governed by a process that is different from the Förster mechanism (J. Phys. Chem. C 2013, 117, 15429–15437. DOI: 10.1021/jp404952x).
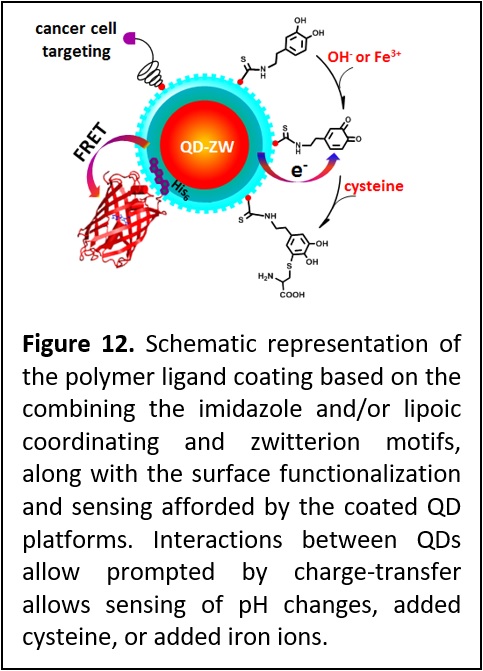
Combining compact polymer ligation with charge transfer based sensing. We have developed a polymeric ligand presenting multiple imidazole and or lipoic acid anchoring groups and a controllable number of zwitterion moieties and applied it to coating QDs and Goal nanostructures alike. The synthesis strategy also permitted the introduction of reactive and biomolecular groups to allow for further conjugation and targeting (Figure 12). We found that QDs capped with these new ligands exhibit excellent long-term colloidal stability over a broad range of pH, to excess electrolyte, in cell growth media and in the presence of natural reducing agents such as glutathione. These QDs are easily self-assembled with full size proteins expressed with a polyhistidine tag via metal-histidine coordination. Additionally, incorporation of amine groups allows covalent coupling of the QDs to the neurotransmitter dopamine. This has yielded redox active QD platforms that were used to track pH changes, sensing interactions with Fe ions and cysteine. Finally, we found that QDs cap exchanged with folic acid-functionalized ligands could effectively target cancer cells, where folate receptor-mediated endocytosis of QDs into living cells (J. Am. Chem. Soc. 2015, 137, 14158–14172. DOI: 10.1021/jacs.5b08915).